
2025年2月26日,88858cc永利官网藥理學系付琴團隊在Redox Biology發表了題為“PDE4D inhibition ameliorates cardiac hypertrophy and heart failure by activating mitophagy”的研究論文,該研究揭示了β-AR持續激活可通過cAMP-PKA上調PDE4D表達,負反饋抑制cAMP-PKA信号通路,導緻心功能下降和線粒體自噬受損,促進心肌肥厚和心力衰竭發生;而PDE4B表達水平不受β-AR激活影響,相反對β-AR持續激活具有保護作用;提出靶向抑制PDE4D可改善心髒功能和心髒重構保護心力衰竭的治療新思路。
心力衰竭是許多心血管疾病的最終階段,引發了嚴重的公共衛生和社會經濟問題。在心力衰竭發生時,交感神經系統過度激活,釋放大量兒茶酚胺類物質激活β-腎上腺素能受體(β-AR)。在初期β-AR激活通過增加環磷酸腺苷(cAMP)水平增強心髒收縮力,以維持心髒的泵血功能;但長期過度激活β-AR會經cAMP-PKA通路促進心肌肥大等心肌重塑過程,導緻心髒功能下降。然而随着長期交感神經興奮心肌細胞β-AR密度下調,cAMP産生不足,心髒重構并未随之緩解,而是進一步惡化加重心衰。
細胞内cAMP水平受磷酸二酯酶(Phosphodiesterases,PDEs)降解調控,而PDE4亞型家族中PDE4B和PDE4D對心肌細胞cAMP降解尤為重要。有觀點認為增加心髒PDE4表達可促進cAMP降解進而改善心髒重構,但也有可能由于cAMP水平降低抑制心髒收縮功能。雖然PDE4抑制劑已經在臨床應用于慢性阻塞性肺疾病,特發性肺纖維化以及銀屑病,然而尚不明确PDE4抑制劑在心衰中的作用及機制。
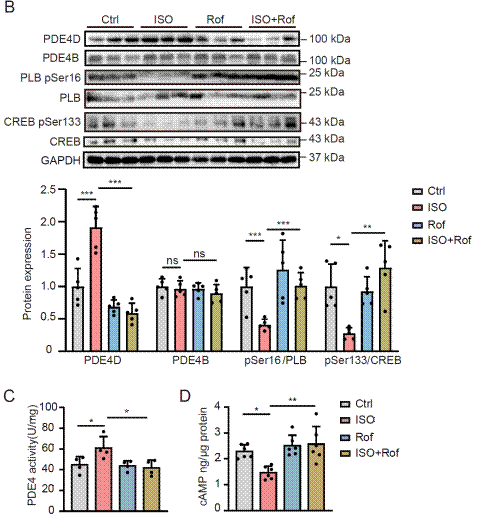
研究發現異丙腎上腺素(ISO)誘導的心力衰竭小鼠心髒PDE4D表達水平顯著上調,而PDE4B表達水平沒有改變,同時PDE4活性增加,而cAMP水平及PKA底物受磷蛋白PLB和CREB磷酸化水平相應下降。PDE4抑制劑Roflumilast在發揮保護ISO誘導的心力衰竭作用中同時抑制PDE4D表達而對于PDE4B沒有影響,提示PDE4D可能參與了ISO誘導心力衰竭的發生。
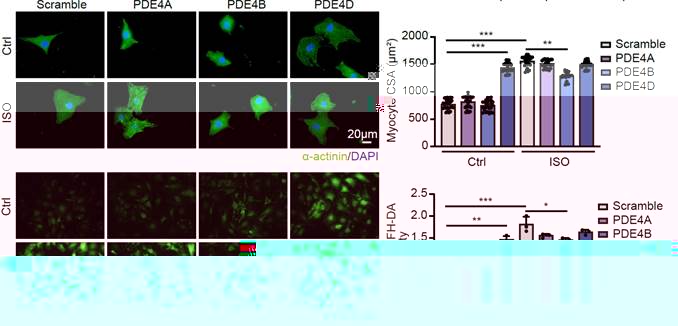
細胞實驗發現ISO通過PKA途徑誘導PDE4D表達,而在心肌細胞過表達PDE4D可直接誘導心肌細胞肥大和氧化應激損傷,PDE4B過表達卻對ISO誘導的心肌細胞損傷有保護作用。
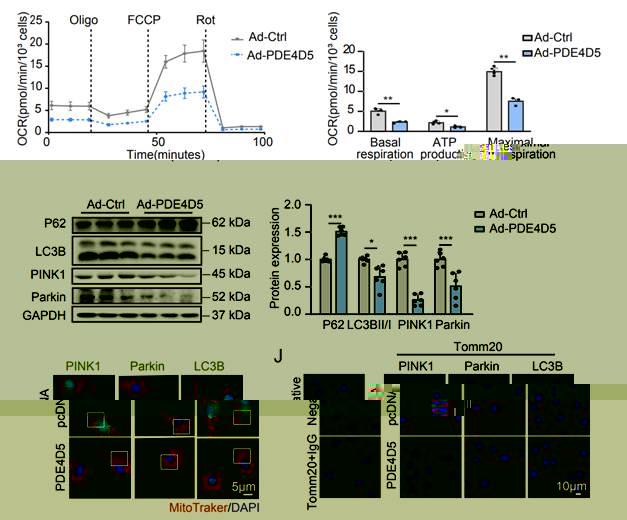
進一步研究發現,在心力衰竭患者心髒PDE4D5表達顯著上調,PDE4D5通過抑制PINK1-Parkin通路介導的線粒體自噬誘導線粒體損傷,促進心肌細胞肥大。
機制探讨發現PDE4D通過抑制胞核CREB磷酸化進而抑制SIRT1介導的線粒體自噬,而PDE4B對線粒體自噬無影響,兩者作用的差異性可能與其在細胞内定位不同有關,PDE4B對胞核CREB活性無影響。


作者還構建了PDE4D心髒特異性敲除小鼠,以及腺相關病毒心髒特異性表達PDE4D5進行回補實驗,在主動脈縮窄(TAC)誘導小鼠心力衰竭模型,驗證了心髒特異性抑制PDE4D表達可通過改善線粒體自噬對心力衰竭的保護作用。

綜上,作者提出持續激活β-AR後通過cAMP-PKA-PDE4D-cAMP↓反饋通路,抑制CREB-SIRT1信号通路,損傷線粒體自噬,誘導心肌肥厚和心衰的發生;靶向抑制PDE4D不僅可恢複cAMP水平改善心功能,還可阻斷PDE4D損傷線粒體自噬的作用改善心髒重構,對心衰具有保護作用。作者還分别在ISO和TAC誘導心衰小鼠驗證了PDE4抑制劑Roflumilast對心髒的保護作用,為臨床轉化提供了新策略。

付晶博士、蘇聰平博士生為本文共同第一作者,付琴教授為本文通訊作者。研究獲得了華中科技大學附屬協和醫院黃恺教授、美國加州大學戴維斯分校Yang K. Xiang教授和廣州醫科大學申翺教授等大力支持。該團隊前期深入研究了腎上腺素受體-PDE4在心髒疾病中的調控作用,并揭示了PDE4在糖尿病心功能不全發生中的關鍵機制以及PDE4抑制劑的保護作用,在JBC、Diabetes和JMCC等雜志發表了系列研究成果。該工作得到國家自然科學基金、湖北省自然科學基金、武漢市自然科學基金等項目支持。
原文鍊接:https://doi.org/10.1016/j.redox.2025.103563

 學院官方微信
學院官方微信